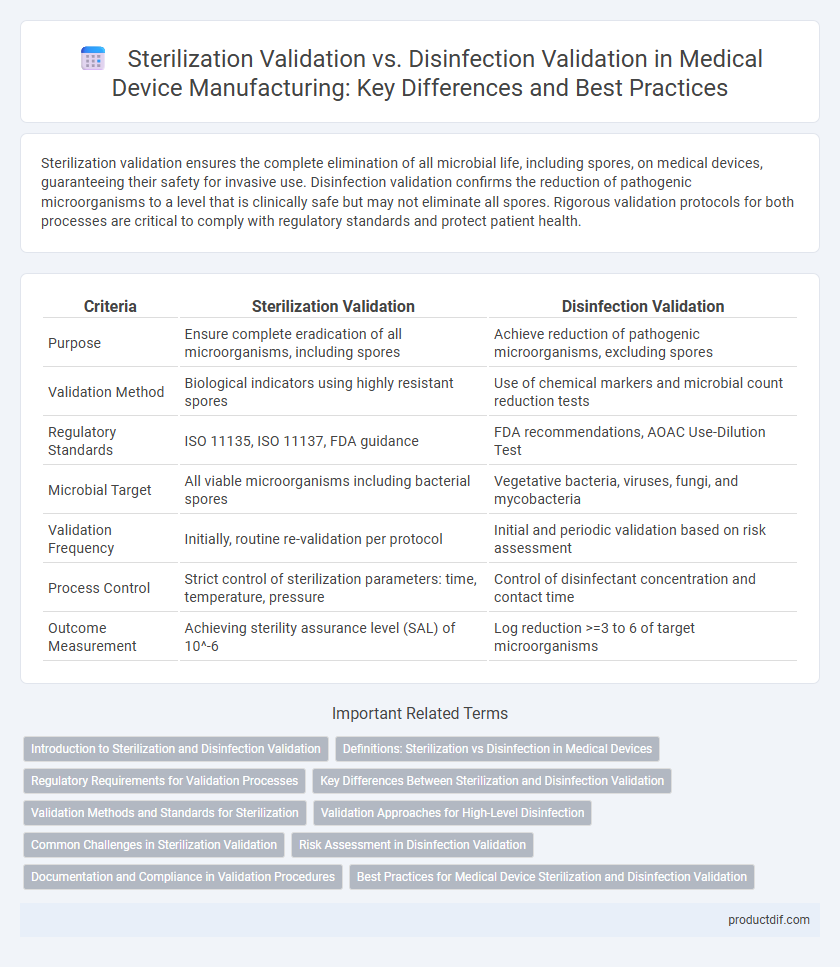
Sterilization validation ensures the complete elimination of all microbial life, including spores, on medical devices, guaranteeing their safety for invasive use. Disinfection validation confirms the reduction of pathogenic microorganisms to a level that is clinically safe but may not eliminate all spores. Rigorous validation protocols for both processes are critical to comply with regulatory standards and protect patient health.
Table of Comparison
| Criteria | Sterilization Validation | Disinfection Validation |
|---|---|---|
| Purpose | Ensure complete eradication of all microorganisms, including spores | Achieve reduction of pathogenic microorganisms, excluding spores |
| Validation Method | Biological indicators using highly resistant spores | Use of chemical markers and microbial count reduction tests |
| Regulatory Standards | ISO 11135, ISO 11137, FDA guidance | FDA recommendations, AOAC Use-Dilution Test |
| Microbial Target | All viable microorganisms including bacterial spores | Vegetative bacteria, viruses, fungi, and mycobacteria |
| Validation Frequency | Initially, routine re-validation per protocol | Initial and periodic validation based on risk assessment |
| Process Control | Strict control of sterilization parameters: time, temperature, pressure | Control of disinfectant concentration and contact time |
| Outcome Measurement | Achieving sterility assurance level (SAL) of 10^-6 | Log reduction >=3 to 6 of target microorganisms |
Introduction to Sterilization and Disinfection Validation
Sterilization validation ensures a medical device is free of all viable microorganisms, including resistant bacterial spores, by evaluating methods like steam, ethylene oxide, or gamma irradiation. Disinfection validation assesses the effectiveness of processes aimed at reducing the number of pathogenic microorganisms to a safe level without necessarily eliminating spores, typically involving chemical disinfectants. Both validations are critical for compliance with ISO 17665 and ISO 17664 standards, ensuring patient safety and regulatory approval.
Definitions: Sterilization vs Disinfection in Medical Devices
Sterilization validation in medical devices ensures the complete elimination of all microorganisms, including bacterial spores, using methods like autoclaving or ethylene oxide sterilization. Disinfection validation targets the reduction of harmful microorganisms to a safe level, but does not guarantee the destruction of spores, commonly achieved through chemical disinfectants such as alcohol or chlorine-based solutions. Understanding the distinct definitions and validation processes of sterilization versus disinfection is crucial for compliance with regulatory standards and ensuring patient safety.
Regulatory Requirements for Validation Processes
Regulatory requirements for sterilization validation mandate comprehensive microbial lethality evidence, ensuring devices achieve a sterility assurance level (SAL) of 10^-6 as per ISO 11135 or ISO 17665 standards. Disinfection validation requires demonstrating effective microbial reduction levels tailored to the device's risk classification, aligning with standards like ANSI/AAMI ST58 but lacks the stringent sterility assurance demanded in sterilization. Both validation processes must include documented protocols, process controls, and routine monitoring to comply with FDA and EMA regulatory guidelines for medical device safety and efficacy.
Key Differences Between Sterilization and Disinfection Validation
Sterilization validation ensures the complete eradication of all microorganisms, including resistant bacterial spores, using methods like steam sterilization or ethylene oxide gas, while disinfection validation focuses on reducing microbial load to safe levels without necessarily eliminating all spores. Sterilization validation requires rigorous biological indicators and physical and chemical parameters for process efficacy, whereas disinfection validation often relies on microbial kill-time studies and concentration monitoring of disinfectants. Understanding these key differences is critical for compliance with regulatory standards such as ISO 11135 for sterilization and CDC guidelines for disinfection in medical device manufacturing.
Validation Methods and Standards for Sterilization
Sterilization validation employs rigorous methods such as biological indicators, chemical indicators, and physical monitoring to ensure the complete eradication of all microorganisms, including spores, adhering to standards like ISO 11135 for ethylene oxide and ISO 17665 for steam sterilization. These protocols require comprehensive process mapping, microbial challenge testing, and verification of sterilant penetration and exposure time to confirm the sterilization efficacy. Disinfection validation, by contrast, focuses on reducing microbial load without necessarily eliminating spores and follows less stringent standards, emphasizing chemical efficacy and contact time evaluations rather than the exhaustive validation frameworks used in sterilization.
Validation Approaches for High-Level Disinfection
Sterilization validation ensures complete destruction of all microorganisms and spores, typically using methods like steam sterilization, ethylene oxide, or hydrogen peroxide plasma. High-level disinfection validation focuses on eliminating all pathogenic microorganisms except bacterial spores, using agents such as glutaraldehyde or ortho-phthalaldehyde, relying on chemical exposure and contact time verification. Validation approaches for high-level disinfection emphasize microbial challenge testing, concentration monitoring, and process parameters to confirm efficacy without reaching sterilization levels.
Common Challenges in Sterilization Validation
Sterilization validation faces common challenges such as ensuring consistent microbial lethality across complex device surfaces and materials, validating sterilization cycles under varied load configurations, and effectively monitoring critical parameters like temperature, pressure, and exposure time. Achieving reproducible results while balancing device integrity and sterility assurance levels (SAL) remains complex, requiring rigorous process design and comprehensive biological indicator testing. Regulatory compliance demands thorough documentation and robust risk assessment to address potential validation failures and process variabilities.
Risk Assessment in Disinfection Validation
Risk assessment in disinfection validation critically evaluates microbial reduction levels against potential contamination risks to ensure patient safety in medical device usage. Unlike sterilization validation, which aims for complete microbial elimination, disinfection validation balances efficacy with material compatibility and operational feasibility. Implementing a thorough risk assessment guides process parameters and controls, optimizing disinfection protocols to minimize infection hazards without compromising device integrity.
Documentation and Compliance in Validation Procedures
Sterilization validation requires comprehensive documentation of process parameters, biological indicators, and equipment calibration to ensure complete microbial elimination, meeting strict regulatory compliance such as ISO 11135 or ISO 17665 standards. Disinfection validation focuses on verifying the efficacy of chemical or thermal processes against specific pathogens, with documentation emphasizing agent concentration, contact time, and efficacy testing per EPA or FDA guidelines. Both validation processes demand rigorous record-keeping to demonstrate adherence to regulatory frameworks, ensuring patient safety and device traceability throughout the medical device lifecycle.
Best Practices for Medical Device Sterilization and Disinfection Validation
Sterilization validation ensures the complete elimination of all microbial life on medical devices, critical for devices entering sterile fields, by rigorously testing methods such as steam autoclaving, ethylene oxide, and hydrogen peroxide plasma. Disinfection validation targets the reduction of pathogenic microorganisms to safe levels using chemical agents or thermal processes, appropriate for semi-critical devices that contact mucous membranes. Best practices include selecting validation methods based on device material compatibility, adhering to recognized standards like ISO 14937 for sterilization and AAMI TIR30 for disinfection, and performing routine efficacy testing to confirm consistent microbial control.
Sterilization Validation vs Disinfection Validation Infographic