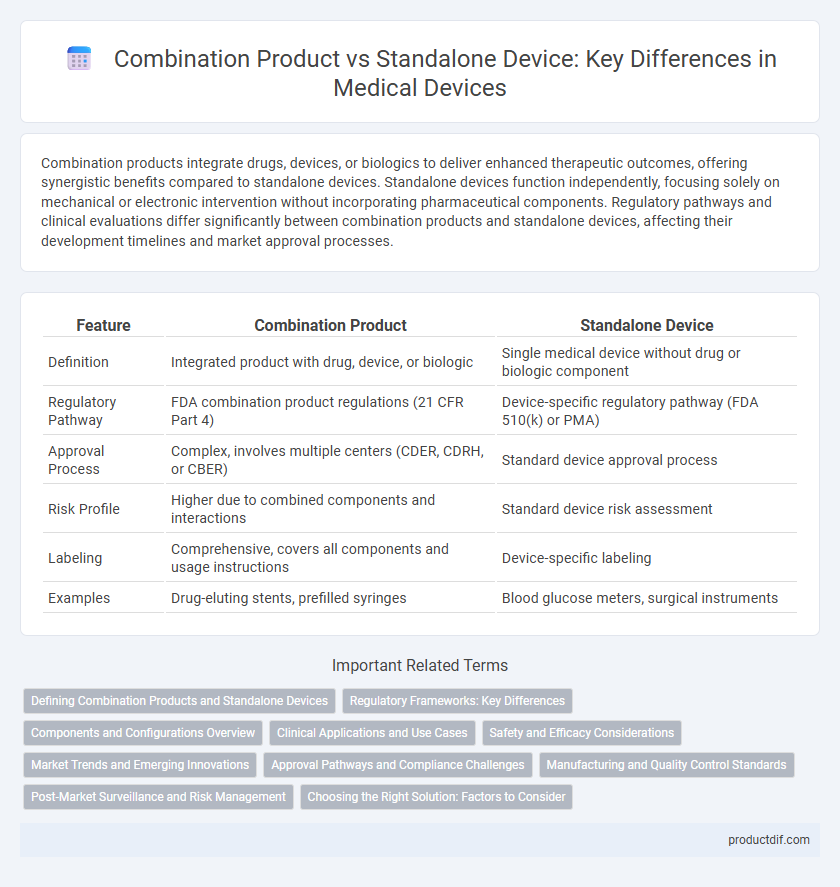
Combination products integrate drugs, devices, or biologics to deliver enhanced therapeutic outcomes, offering synergistic benefits compared to standalone devices. Standalone devices function independently, focusing solely on mechanical or electronic intervention without incorporating pharmaceutical components. Regulatory pathways and clinical evaluations differ significantly between combination products and standalone devices, affecting their development timelines and market approval processes.
Table of Comparison
| Feature | Combination Product | Standalone Device |
|---|---|---|
| Definition | Integrated product with drug, device, or biologic | Single medical device without drug or biologic component |
| Regulatory Pathway | FDA combination product regulations (21 CFR Part 4) | Device-specific regulatory pathway (FDA 510(k) or PMA) |
| Approval Process | Complex, involves multiple centers (CDER, CDRH, or CBER) | Standard device approval process |
| Risk Profile | Higher due to combined components and interactions | Standard device risk assessment |
| Labeling | Comprehensive, covers all components and usage instructions | Device-specific labeling |
| Examples | Drug-eluting stents, prefilled syringes | Blood glucose meters, surgical instruments |
Defining Combination Products and Standalone Devices
Combination products integrate two or more regulated components, such as a drug and a medical device, intended to function as a single entity to enhance therapeutic outcomes. Standalone devices operate independently without relying on drugs or biologics, designed for specific medical purposes like diagnostics or treatment. Regulatory frameworks distinguish these categories to ensure appropriate evaluation and compliance based on their unique characteristics and risk profiles.
Regulatory Frameworks: Key Differences
Combination products are regulated under complex frameworks that integrate requirements for both medical devices and pharmaceutical products, often involving agencies like the FDA's Office of Combination Products, while standalone devices follow device-specific regulations such as FDA's 21 CFR Part 820 Quality System Regulation. Regulatory pathways for combination products demand thorough evaluation of both device safety and drug efficacy, requiring coordinated premarket submissions and labeling controls reflecting dual compliance. In contrast, standalone devices undergo streamlined assessments focusing solely on device performance, biocompatibility, and mechanical integrity under device-centric regulatory standards.
Components and Configurations Overview
Combination products integrate drug, device, and/or biological components into a single entity, requiring coordinated design and regulatory strategies to ensure safety and efficacy. Standalone devices function independently, focusing solely on mechanical or electronic systems without incorporating pharmaceutical elements. Configurations of combination products vary widely, including drug-device, biologic-device, and drug-biologic-device combinations, each demanding specific considerations for component interaction and compatibility.
Clinical Applications and Use Cases
Combination products integrate medical devices with drugs or biologics, enhancing therapeutic outcomes in complex clinical applications such as drug delivery systems and wound care management. Standalone devices, like pacemakers or diagnostic imaging tools, serve specific functions independently, often excelling in monitoring, diagnostics, or mechanical support. Clinical use cases for combination products include controlled drug release and localized treatment, whereas standalone devices are typically utilized for continuous monitoring or structural intervention.
Safety and Efficacy Considerations
Combination products integrate a drug, device, or biological product, which requires comprehensive safety and efficacy evaluations to address potential interactions and compounded risks. Standalone devices undergo focused assessments specific to their mechanical or technological functions, allowing for targeted validation of safety protocols and performance metrics. Regulatory scrutiny for combination products is often more complex, necessitating rigorous clinical testing to ensure combined components do not compromise patient safety or therapeutic effectiveness.
Market Trends and Emerging Innovations
Combination products integrating drug, device, and biological components are experiencing rapid market growth, driven by personalized medicine and targeted therapies. Emerging innovations such as smart drug delivery systems and wearable biosensors enhance functionality beyond standalone devices, offering improved patient outcomes and adherence. Regulatory shifts and increased investment in combination technologies signal a transformative trend in medical device development and commercialization.
Approval Pathways and Compliance Challenges
Combination products, integrating drugs, devices, or biologics, encounter complex FDA approval pathways often requiring coordination between the Center for Drug Evaluation and Research (CDER) and the Center for Devices and Radiological Health (CDRH). Standalone medical devices typically follow a more straightforward regulatory route such as 510(k) clearance or Premarket Approval (PMA), with compliance focused on device-specific standards like ISO 13485 and IEC 60601. Combination products face heightened compliance challenges involving both pharmaceutical Good Manufacturing Practices (GMP) and device Quality System Regulations (QSR), necessitating rigorous cross-disciplinary quality management systems.
Manufacturing and Quality Control Standards
Combination products require integrated manufacturing processes that adhere to both medical device and pharmaceutical quality control standards, ensuring compliance with FDA's Quality System Regulation (QSR) and Current Good Manufacturing Practice (cGMP) guidelines. Standalone devices emphasize strict adherence to ISO 13485 standards for medical device quality management systems, focusing primarily on device-specific design controls and validation procedures. Manufacturing combination products involves complex risk management strategies and cross-disciplinary quality audits to maintain regulatory compliance and product safety throughout the production lifecycle.
Post-Market Surveillance and Risk Management
Combination products require integrated post-market surveillance systems that monitor both the device and drug components, ensuring comprehensive risk management across all constituent parts. Standalone devices typically rely on device-specific vigilance protocols and risk assessment tools to detect and mitigate adverse events. Regulatory agencies emphasize tailored surveillance strategies to address the unique safety profiles and failure modes presented by combination products compared to standalone medical devices.
Choosing the Right Solution: Factors to Consider
Choosing between a combination product and a standalone medical device depends on regulatory requirements, intended use, and patient safety profiles. Combination products, integrating drugs, devices, or biologics, require comprehensive evaluation of component interactions and FDA jurisdictional oversight, whereas standalone devices primarily focus on device-specific performance standards. Understanding clinical benefits, manufacturing complexity, and market access implications is crucial for optimal product strategy and compliance.
Combination product vs Standalone device Infographic