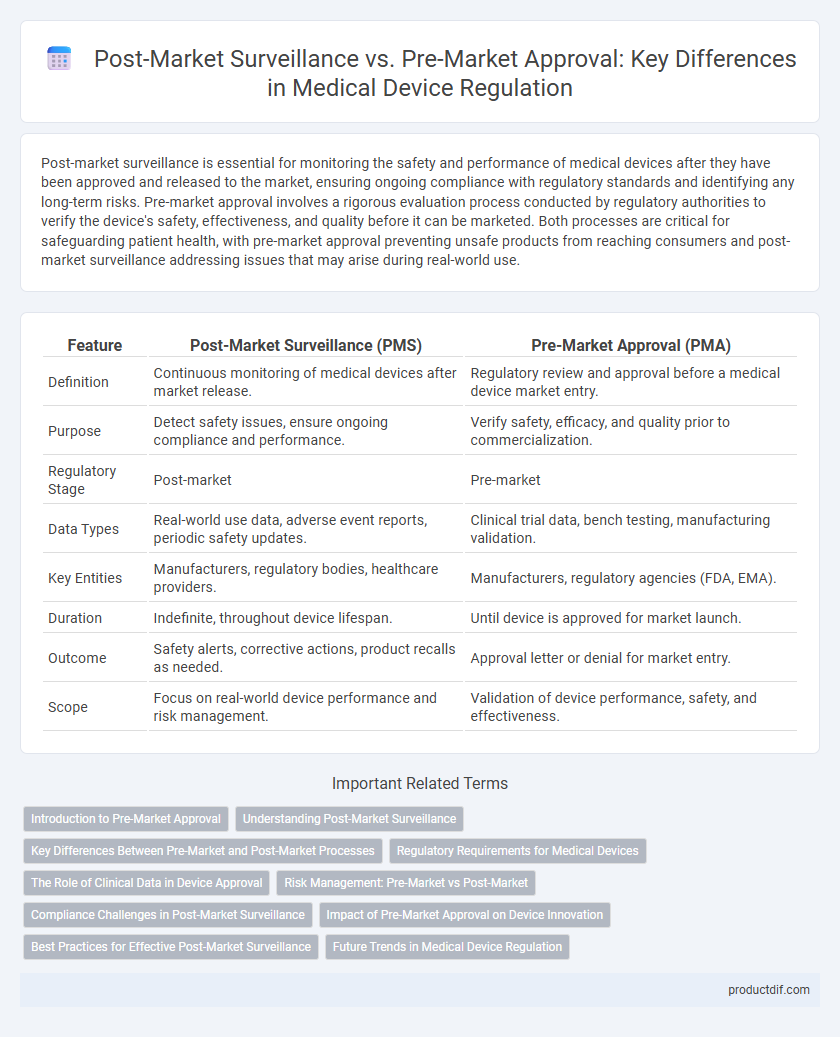
Post-market surveillance is essential for monitoring the safety and performance of medical devices after they have been approved and released to the market, ensuring ongoing compliance with regulatory standards and identifying any long-term risks. Pre-market approval involves a rigorous evaluation process conducted by regulatory authorities to verify the device's safety, effectiveness, and quality before it can be marketed. Both processes are critical for safeguarding patient health, with pre-market approval preventing unsafe products from reaching consumers and post-market surveillance addressing issues that may arise during real-world use.
Table of Comparison
| Feature | Post-Market Surveillance (PMS) | Pre-Market Approval (PMA) |
|---|---|---|
| Definition | Continuous monitoring of medical devices after market release. | Regulatory review and approval before a medical device market entry. |
| Purpose | Detect safety issues, ensure ongoing compliance and performance. | Verify safety, efficacy, and quality prior to commercialization. |
| Regulatory Stage | Post-market | Pre-market |
| Data Types | Real-world use data, adverse event reports, periodic safety updates. | Clinical trial data, bench testing, manufacturing validation. |
| Key Entities | Manufacturers, regulatory bodies, healthcare providers. | Manufacturers, regulatory agencies (FDA, EMA). |
| Duration | Indefinite, throughout device lifespan. | Until device is approved for market launch. |
| Outcome | Safety alerts, corrective actions, product recalls as needed. | Approval letter or denial for market entry. |
| Scope | Focus on real-world device performance and risk management. | Validation of device performance, safety, and effectiveness. |
Introduction to Pre-Market Approval
Pre-Market Approval (PMA) is a rigorous evaluation process required by regulatory authorities, such as the FDA, to ensure the safety and effectiveness of high-risk medical devices before they enter the market. This process involves detailed clinical data analysis, manufacturing controls assessment, and risk-benefit evaluation to verify that the device meets all regulatory standards. PMA plays a critical role in minimizing patient risks by thoroughly vetting novel medical technologies prior to widespread clinical use.
Understanding Post-Market Surveillance
Post-market surveillance (PMS) is a critical phase in the lifecycle of a medical device that involves continuous monitoring of the device's safety and performance after it has received regulatory approval and entered the market. This process collects real-world data and adverse event reports to identify potential risks or defects not detected during pre-market clinical trials, ensuring ongoing compliance with regulatory standards such as the FDA's Quality System Regulation or the EU Medical Device Regulation. Effective PMS enables manufacturers to implement timely corrective actions, enhance device safety, and maintain public health confidence throughout the device's commercial lifespan.
Key Differences Between Pre-Market and Post-Market Processes
Pre-market approval for medical devices involves rigorous evaluation of safety, effectiveness, and compliance with regulatory standards before the device enters the market, typically including clinical trials and detailed documentation submission to authorities such as the FDA. Post-market surveillance, by contrast, continuously monitors the device's performance and adverse events after commercialization, using real-world data, user feedback, and reporting systems to identify risks and ensure ongoing safety. Key differences lie in timing, objectives, and data sources: pre-market processes emphasize controlled testing and initial risk assessment, whereas post-market activities focus on long-term safety monitoring and risk mitigation in diverse real-world environments.
Regulatory Requirements for Medical Devices
Post-Market Surveillance involves continuous monitoring of medical devices after approval to detect any unexpected issues, ensuring ongoing compliance with safety and performance standards. Pre-Market Approval requires rigorous clinical evaluation and documentation to demonstrate device safety and efficacy before market entry, following regulatory standards like FDA's 21 CFR Part 814. Both processes are essential for regulatory compliance, with Pre-Market Approval focusing on initial risk assessment and Post-Market Surveillance ensuring long-term device safety and effectiveness.
The Role of Clinical Data in Device Approval
Clinical data plays a critical role in both post-market surveillance and pre-market approval of medical devices by providing evidence of safety and effectiveness. During pre-market approval, clinical trials generate essential data to demonstrate device performance and support regulatory submission. Post-market surveillance uses real-world clinical data to monitor device performance, identify adverse events, and ensure ongoing compliance with safety standards.
Risk Management: Pre-Market vs Post-Market
Risk management in medical devices involves comprehensive hazard identification and mitigation both during pre-market approval and post-market surveillance. Pre-market risk management focuses on analyzing design and manufacturing risks to ensure device safety before commercialization, while post-market risk management continuously monitors real-world device performance to identify emerging risks and implement corrective actions. Effective integration of pre-market and post-market data enhances overall patient safety and regulatory compliance throughout the device lifecycle.
Compliance Challenges in Post-Market Surveillance
Post-market surveillance (PMS) presents significant compliance challenges distinct from pre-market approval (PMA), as manufacturers must continuously monitor device performance and adverse events in real-world use. Regulatory frameworks like the FDA's 21 CFR Part 822 and the EU MDR require comprehensive data collection, risk management updates, and timely reporting, which demand robust systems and resources. Ensuring ongoing compliance involves integrating real-time data analytics, managing field safety corrective actions, and addressing variability in device usage across diverse patient populations.
Impact of Pre-Market Approval on Device Innovation
Pre-Market Approval (PMA) imposes rigorous testing and compliance standards that can extend the development timeline and increase costs for medical devices, potentially slowing innovation. However, these stringent requirements ensure higher safety and efficacy, fostering greater trust among healthcare providers and patients. Balancing robust pre-market scrutiny with adaptive regulatory pathways can stimulate innovative device design while maintaining patient protection.
Best Practices for Effective Post-Market Surveillance
Post-market surveillance (PMS) in medical devices involves continuous monitoring of device performance and safety after regulatory pre-market approval to ensure ongoing compliance and patient safety. Best practices for effective PMS include implementing robust risk management systems, real-time adverse event reporting, and integrating predictive analytics to identify potential device failures early. Collaboration between manufacturers, healthcare providers, and regulatory bodies enhances data quality and supports proactive corrective actions.
Future Trends in Medical Device Regulation
Future trends in medical device regulation emphasize enhanced integration of real-world data from post-market surveillance to support dynamic pre-market approval processes. Advanced AI algorithms and digital health technologies promote continuous monitoring, enabling faster identification of safety issues and more adaptive regulatory responses. Regulatory frameworks are shifting toward harmonized international standards, facilitating global device approvals while ensuring patient safety through robust lifecycle management.
Post-Market Surveillance vs Pre-Market Approval Infographic