CE Mark certification indicates that a medical device complies with European Union safety, health, and environmental protection requirements, allowing it to be marketed within the European Economic Area. FDA approval signifies that a medical device meets the United States Food and Drug Administration's rigorous standards for safety, efficacy, and performance before entering the U.S. market. Understanding the differences between CE Mark and FDA approval is crucial for manufacturers aiming to access global markets and ensure regulatory compliance.
Table of Comparison
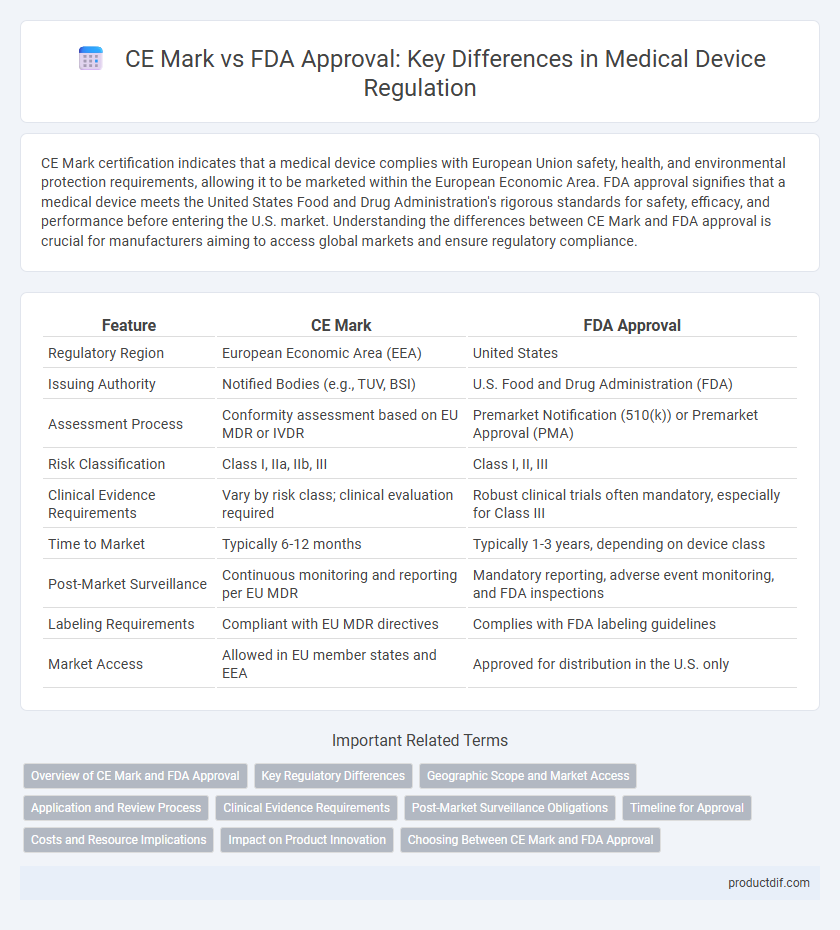
| Feature | CE Mark | FDA Approval |
|---|---|---|
| Regulatory Region | European Economic Area (EEA) | United States |
| Issuing Authority | Notified Bodies (e.g., TUV, BSI) | U.S. Food and Drug Administration (FDA) |
| Assessment Process | Conformity assessment based on EU MDR or IVDR | Premarket Notification (510(k)) or Premarket Approval (PMA) |
| Risk Classification | Class I, IIa, IIb, III | Class I, II, III |
| Clinical Evidence Requirements | Vary by risk class; clinical evaluation required | Robust clinical trials often mandatory, especially for Class III |
| Time to Market | Typically 6-12 months | Typically 1-3 years, depending on device class |
| Post-Market Surveillance | Continuous monitoring and reporting per EU MDR | Mandatory reporting, adverse event monitoring, and FDA inspections |
| Labeling Requirements | Compliant with EU MDR directives | Complies with FDA labeling guidelines |
| Market Access | Allowed in EU member states and EEA | Approved for distribution in the U.S. only |
Overview of CE Mark and FDA Approval
The CE Mark certifies that a medical device complies with European Union safety, health, and environmental requirements, allowing market access across EU member states. FDA Approval involves a rigorous evaluation process by the U.S. Food and Drug Administration to ensure medical devices meet safety and efficacy standards specific to the United States. Both certifications require comprehensive clinical data, risk assessments, and quality management system compliance but differ in regulatory pathways and geographic scope.
Key Regulatory Differences
CE Mark certification indicates compliance with European Union medical device regulations, primarily emphasizing product safety and performance through conformity assessment processes conducted by notified bodies. FDA approval in the United States involves a rigorous evaluation of safety and effectiveness based on clinical data, requiring premarket notification [510(k)] or premarket approval (PMA) pathways depending on the device class. Key regulatory differences include the CE Mark's focus on self-certification with post-market surveillance, whereas FDA approval demands extensive premarket clinical evidence and formal agency clearance before market entry.
Geographic Scope and Market Access
CE Mark certification enables medical devices to access the European Economic Area (EEA), facilitating market entry across over 30 countries including EU member states, Norway, Iceland, and Liechtenstein. FDA approval governs the U.S. market, ensuring compliance with stringent safety and efficacy standards specific to American regulations. Medical device manufacturers must navigate differing regional requirements, as CE Mark emphasizes conformity with EU directives while FDA approval demands rigorous clinical validation and post-market surveillance in the United States.
Application and Review Process
CE Mark application for medical devices involves conformity assessment by a notified body, emphasizing compliance with the EU Medical Device Regulation (MDR) or In Vitro Diagnostic Regulation (IVDR), with a review process often shorter than FDA approval. FDA approval requires submission of a premarket notification (510(k)) or premarket approval (PMA), involving rigorous clinical data and an in-depth evaluation by the FDA Center for Devices and Radiological Health (CDRH). While CE Mark prioritizes product safety and performance compliance across European Economic Area (EEA), FDA approval demands proof of safety and effectiveness specific to the U.S. market, influencing timelines and documentation requirements.
Clinical Evidence Requirements
CE Mark clinical evidence requirements emphasize demonstrating safety and performance through conformity with EU Medical Device Regulation (MDR) standards, including clinical evaluations supported by relevant data such as clinical investigations and literature reviews. FDA approval demands more rigorous clinical evidence, typically requiring substantial data from well-controlled clinical trials that prove safety and efficacy under Food and Drug Administration guidelines. Both regulatory pathways prioritize patient safety but differ in the depth, scope, and explicitness of clinical data requirements during the premarket evaluation process.
Post-Market Surveillance Obligations
CE Mark post-market surveillance requires continuous vigilance through periodic safety update reports (PSUR) and proactive risk management per EU MDR 2017/745 standards. FDA approval mandates rigorous post-market surveillance including Medical Device Reporting (MDR), post-approval studies, and adverse event monitoring through the FDA's MedWatch system. Compliance with both frameworks ensures ongoing device safety and effectiveness by systematically collecting and analyzing real-world performance data.
Timeline for Approval
The CE Mark typically requires a shorter approval timeline, often ranging from a few months to a year, as it relies on conformity assessment by notified bodies under the European Union's Medical Device Regulation (MDR). In contrast, FDA approval, especially through the Premarket Approval (PMA) process, can take several years due to rigorous clinical trial requirements and thorough evaluation by the Center for Devices and Radiological Health (CDRH). The 510(k) pathway offers a faster FDA review, usually 3 to 6 months, but applies only to devices demonstrating substantial equivalence to legally marketed predicates.
Costs and Resource Implications
CE Mark certification typically incurs lower upfront costs and faster market access within the European Economic Area, but requires ongoing compliance with regional regulations. FDA approval involves higher initial expenses due to rigorous premarket submissions and clinical trials, demanding extensive documentation and resource allocation for U.S. market entry. Resource implications for FDA processes often include dedicated regulatory teams, while CE Mark processes allow more flexibility through notified bodies and decentralized conformity assessments.
Impact on Product Innovation
CE Mark facilitates faster market entry in the European Union, enabling medical device manufacturers to accelerate product innovation cycles and respond quickly to emerging healthcare needs. In contrast, FDA approval in the United States demands rigorous clinical data and longer review periods, which can delay time-to-market but ensures high safety and efficacy standards that drive incremental innovation. The differing regulatory pathways influence strategic planning for R&D investments, with CE Mark favoring rapid prototyping and FDA approval promoting thorough validation processes.
Choosing Between CE Mark and FDA Approval
Choosing between CE Mark and FDA approval depends on the target market and regulatory standards for medical devices. CE Mark indicates compliance with European Union directives, allowing access to the European Economic Area, while FDA approval is mandatory for marketing devices in the United States, emphasizing stringent safety and efficacy criteria. Understanding the device classification, regulatory pathways, and timeframes for each jurisdiction is crucial for efficient market entry and ensuring patient safety.
CE Mark vs FDA Approval Infographic